
Displasia Epifisária Múltipla
A Displasia Epifisária Múltipla (DEM) corresponde a um grupo de condições que afetam a cartilagem e o crescimento ósseo, principalmente das extremidades dos ossos longos dos braços e pernas (epífises). A DEM pode ser distinguida pelo seu padrão hereditário, dominante (DEMd) ou recessivo (DEMr).
A DEMd ocorre em aproximadamente 1 em 10.000 nados vivos, enquanto que a incidência da DEMr é desconhecida. Ambas as formas desta displasia óssea podem, na realidade, ser mais comuns porque algumas pessoas com sintomas ligeiros nunca são diagnosticadas1.
O que é a DEM1,2?
A DEM, também conhecida como doença de Fairbank, é uma doença genética rara (forma dominante: 1 em cada 10.000 nascimentos) que afeta as extremidades dos ossos em crescimento. Os ossos longos alongam-se normalmente através da expansão da cartilagem na placa de crescimento (placa epifisária) perto das suas extremidades. À medida que se expande para fora da placa de crescimento, a cartilagem mineraliza-se e endurece, transformando-se em osso (ossificação). Na DEM, este processo é defeituoso.
O que causa a DEM2,3,4,5?
A DEM pode ter diferentes padrões de hereditariedade. Pode, ser herdada num padrão autossómico dominante (uma cópia do gene alterado em cada célula é suficiente para a causar). Em alguns casos a mutação é herdada de um dos pais afectados. Outros casos podem resultar de novas mutações no gene, sem historial da doença na família. A sua forma dominante é a mais comum2:
-
Mutações nos genes COMP e MATN3 podem causar DEMd2. Os genes COMP e MATN3 fornecem instruções para produzir proteínas que se encontram nos espaços entre as células formadoras de cartilagem (condrócitos). Estas proteínas interagem entre si e desempenham um papel importante na formação de cartilagens e ossos e a sua ausência leva à formação de cartilagem anómala, causando os problemas esqueléticos característicos da DEMd2.
-
Mutações nos genes COL9A1, COL9A2, e COL9A3 são mais raras (encontradas em menos de 5% dos indivíduos com DEMd) mas também pode causar DEMd3. Estes genes fornecem instruções para fazer uma proteína chamada colagénio tipo IX. No entanto, não se sabe ao certo como causam os sinais e sintomas desta condição. A investigação sugere que as mutações nestes genes poderão causar a acumulação de colagénio tipo IX no interior da célula ou interagir anormalmente com outros componentes da cartilagem3.
Pode também ser herdada num padrão autossómico recessivo. Neste caso, os pais de um indivíduo transportam, cada um, uma cópia do gene mutado, mas não mostram, habitualmente, sinais e sintomas da doença4.
Mutações no gene SLC26A2 causam DEMr5. Este gene fornece instruções para produzir uma proteína essencial para o desenvolvimento normal da cartilagem e para a sua conversão em osso e as mutações neste gene resultam nos problemas esqueléticos característicos da DEMr5.

Fig. 1. Padrões de hereditariedade dominante e recessiva.
Características Clínicas e Radiológicas6,7,8
Tanto o tipo dominante como o recessivo têm sinais e sintomas relativamente ligeiros, incluindo dores nas articulações que afectam mais frequentemente ancas e joelhos, artrite precoce, e causam um andar bamboleante. Embora algumas pessoas com DEM tenham uma ligeira baixa estatura em idade adulta, a maioria tem uma estatura normal. Características mais comuns:
-
Problemas da anca devido a desalinhamento, subluxação ou doença de Perthes*
-
Problemas nos joelhos devido a desalinhamento (genu valgum ou varum)
-
Problemas nos tornozelo devido a desalinhamento
-
Rótula de camada dupla (patella)
-
Artrite precoce na anca, joelhos e ombros, que pode ocorrer na faixa dos 20 aos 30 anos de idade
-
Necrose avascular das cabeças femorais.
*Doença de Perthes é uma condição que ocorre nas crianças e que se caracteriza por uma perda momentânea do fluxo sanguíneo na região da anca, com consequente morte dos tecidos da cabeça do fémur. A área afetada apresenta-se muito inflamada e irritada.
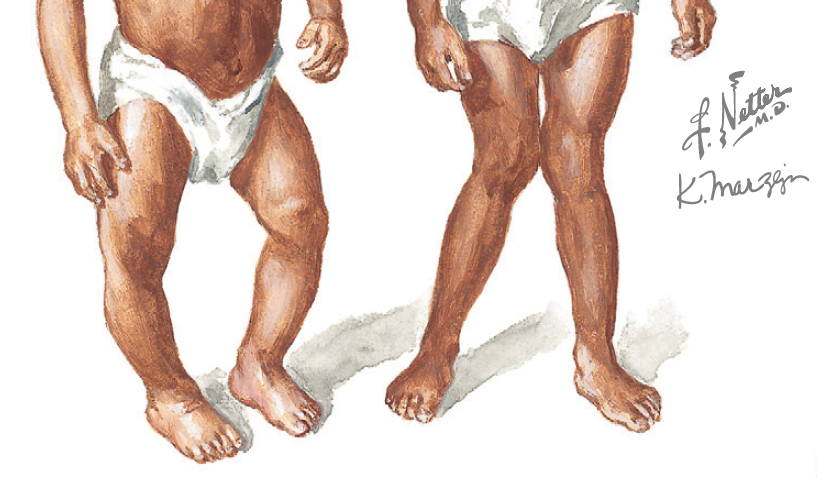
Fig. 1. Genu Varum e Valgum. Créditos: F. Netter
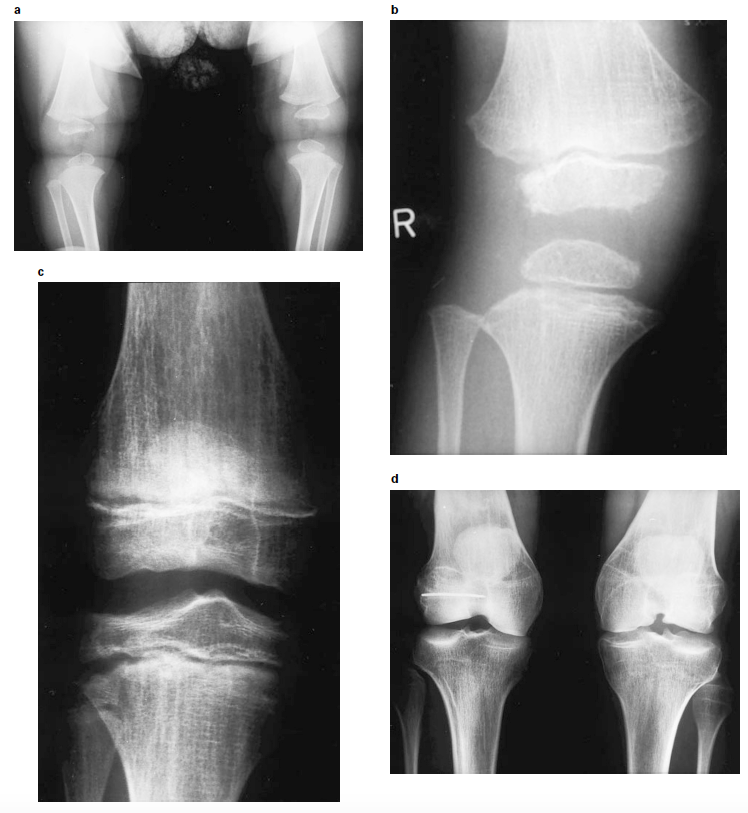
Fig. 2. Radiografias dos joelhos em diferentes idades. a) Joelho de bebé de 8 meses com pequenas epífises com contornos irregulares. (b) joelho aos 5 anos de idade mostra ausência de ossificação da epífise fibular, epífises tibiais e femorais com contornos irregulares (mais pronunciadas na epífise femoral distal), metáfise tibial anómala com defeito de ossificação lateral e borda translúcida e irregular. A metáfise femoral é também irregular com estrias longitudinais. (c) joelho aos 14 anos de idade demonstra epífises achatadas e estrias metafisárias longitudinais, mais pronunciadas na metáfise distal do fémur. (d) Displasia e artrose dos joelhos no indivíduo aos 29 anos de idade. Créditos: European Journal of Human Genetics
O tipo recessivo distingue-se do dominante, apresentando características distintas. Cerca de 50 por cento dos indivíduos com DEMr apresentam pelo menos uma das seguintes características à nascença4:
-
Malformações das mãos, pés e joelhos e curvatura anómala da coluna vertebral (escoliose).
-
Pé torcido para dentro e para cima (pé torto)
-
Abertura no céu da boca (palato fendido)
-
Curvatura invulgar dos dedos das mãos ou dos pés (clinodactilia)
-
Inchaço da orelha.
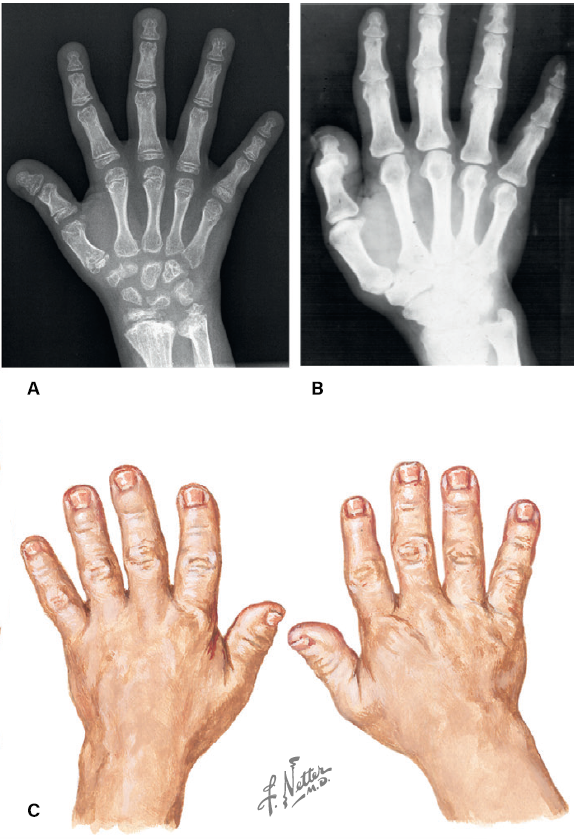
Fig. 3. Aos 5 anos, ossificação retardada das epífises na mão e no pulso (A).
Aos 42 anos, superfícies de articulação irregulares e estreitas (B).
Aos 40 anos, mãos largas com "polegares de oleiro" curtos, largos e achatados (C). Créditos: F. Netter
Diagnóstico8,9
Um diagnóstico preciso requer um exame físico pormenorizado, um levantamento radiográfico completo do esqueleto (as radiografias demonstram ossificação irregular e retardada em múltiplas epífises) e uma história familiar detalhada. O diagnóstico é frequente durante a infância, contudo, alguns casos ligeiros podem não ser diagnosticados até à idade adulta.
- Diagnóstico diferencial: Displasia espondiloepifisária congénita; Mucopolissacaridoses; Hipotiroidismo congénito.
É possível alcançar o diagnóstico através de testes genéticos. O Registo de Testes Genéticos fornece informações sobre os testes genéticos para esta condição. Quando é feito um diagnóstico clínico de DEM deve ser oferecido aconselhamento genético.
Tratamento1,10,11,12
Geralmente, todas as displasias ósseas merecem uma atenção multidisciplinar. A avaliação regular por um ortopedista, geneticista, pediatra, dentista, neurologista e fisioterapeuta proporcionará um tratamento mais abrangente e adequado.
O tratamento varia em função das condições ortopédicas associadas que a pessoa afetada apresenta e pode incluir:
-
Fisioterapia
-
Cirurgia de realinhamento da anca
-
Crescimento guiado das extremidades inferiores (hemiepifisiodese) para ajudar a corrigir deformidades
-
Osteotomias para deformidades graves das extremidades inferiores
-
Excisão de patela no caso de ocorrer uma camada dupla e estiver a causar dor
-
Substituição total das articulações (artroplastia) da anca, joelhos e ombros.
Embora a DEM seja uma doença da infância, raramente é suficientemente grave para requerer uma intervenção cirúrgica nas duas primeiras décadas de vida. Deve por isso haver algum cuidado com o excesso de tratamento.
As pessoas afetadas devem abster-se de desportos e atividades de alto impacto, tais como jogging. Todavia, nadar e andar de bicicleta são desportos sugeridos pois não infligem peso nas articulações.
Bibliografia
- Dahlqvist J, et al. Multiple epiphyseal dysplasia. Acta Orthop 2009;80(6):711–715.
- Briggs MD, et al. Multiple Epiphyseal Dysplasia, Autosomal Dominant. 2003 Jan 8. In: Adam MP, et al. GeneReviews® . Seattle (WA): University of Washington. Disponível em http://www.ncbi.nlm.nih.gov/books/NBK1123/
- Jackson GC, et al. Type IX collagen gene mutations can result in multiple epiphyseal dysplasia that is associated with osteochondritis dissecans and a mild myopathy. Am J Med Genet A. 2010 Apr;152A(4):863-9. doi: 10.1002/ajmg.a.33240
- Ballhausen D, et al. Recessive multiple epiphyseal dysplasia (rMED): phenotype delineation in eighteen homozygotes for DTDST mutation R279W. J Med Genet. 2003 Jan;40(1):65-71
- Rossi A, Superti-Furga A. Mutations in the diastrophic dysplasia sulfate transporter (DTDST) gene (SLC26A2): 22 novel mutations, mutation review, associated skeletal phenotypes, and diagnostic relevance. Hum Mutat. 2001 Mar;17(3):159-71.
- Mäkitie O, et al.: Clinical and radiographic findings in multiple epiphyseal dysplasia caused by MATN3 mutations: Description of 12 patients. Am J Med Genet A 2004;125A(3):278–284.
- Mortier GR, et al. Clinical and radiographic features of multiple epiphyseal dysplasia not linked to the COMP or type IX collagen genes. Eur J Hum Genet 2001;9(8):606-12.
- Unger S, et al. Multiple epiphyseal dysplasia: clinical and radiographic features, differential diagnosis and molecular basis,Best Practice & Research Clinical Rheumatology,Volume 22, Issue 1,2008,Pages 19-32,ISSN 1521-6942,https://doi.org/10.1016/j.berh.2007.11.009.
- Ingram RR. Early diagnosis of multiple epiphyseal dysplasia. J Pediatr Orthop 1992;12(2):241-4.
- Akhmedov B, et al. Reliability of lower-limb alignment measurements in patients with multiple epiphyseal dysplasia. Clin Orthop Relat Res 2012;470(12):3566–3576.
- Sebik A, et al: The orthopaedic aspects of multiple epiphyseal dysplasia. Int Orthop 1998;22(6):417–421.
- Li LY, et al. Clinical features and treatment of the hip in multiple epiphyseal dysplasia in childhood. Orthopedics 2011;34(5):352.
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui
Partilhe a sua história
Gostaria de partilhar a sua experiência? Pode fazê-lo na primeira pessoa ou enquanto mãe, pai ou cuidador. O seu testemunho pode ajudar a informar, sensibilizar e inspirar outras pessoas.
partilhar a minha história