Displasia Metatrópica
A displasia metatrópica é uma condição rara que afeta o esqueleto descrita pela primeira vez em 1966. Caracteriza-se por apresentar um tronco longo e membros curtos na infância, seguidos de uma cifoscoliose grave e progressiva, causando uma inversão de proporções durante a infância (tronco curto e membros longos) e uma estatura final curta na idade adulta.
O que é a Displasia Metatrópica?
A displasia metatrópica é uma displasia espondiloepimetafisária rara transmitida geneticamente de forma autossómica dominante. O que significa que um gene dominante de um dos progenitores é suficiente para causar a condição. A sua prevalência exata é desconhecida e apenas foram descritos 81 casos na literatura científica, até à data.
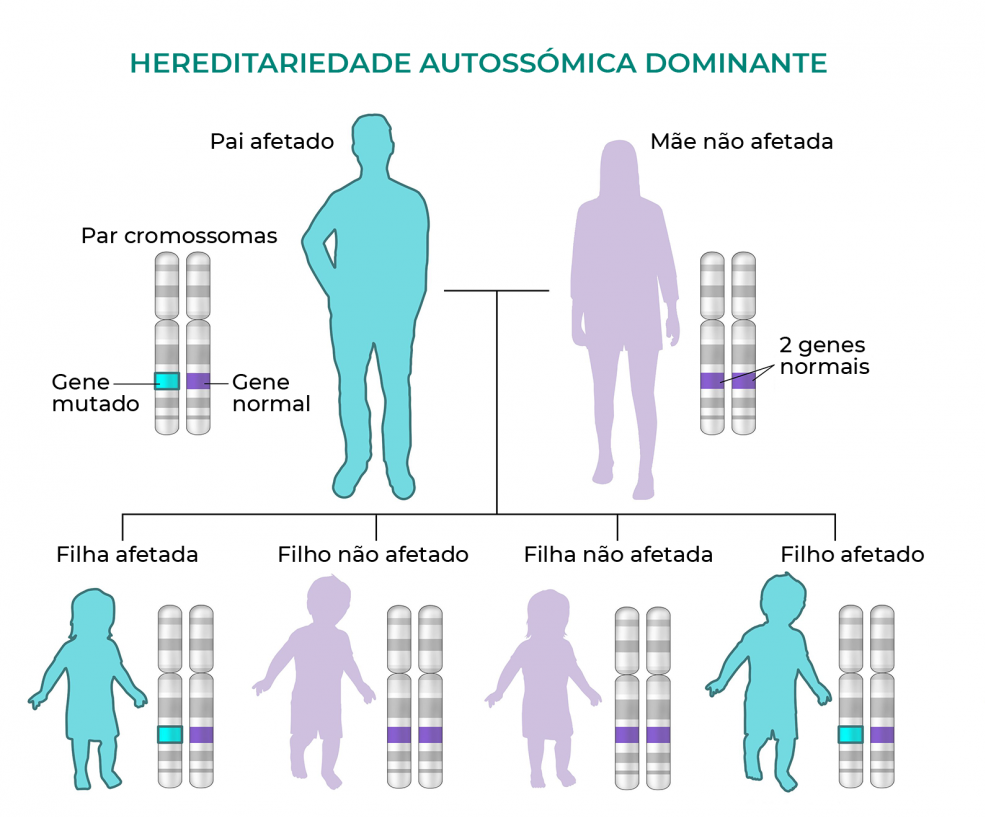
Hereditariedade
A displasia metatrópica é considerada uma condição autossómica dominante porque uma cópia mutada do gene em cada célula é suficiente para a causar. No entanto, maioria dos casos são causados por novas mutações no gene e ocorrem em pessoas sem historial da condição na família (mutações de novo).
O termo metatrópico vem da palavra grega metatropos que significa "mudar de forma", referindo-se à principal característica clínica da condição: uma mudança do período recém-nascido, quando apresenta tronco longo e membros curtos, para a infância, quando, devido a alterações progressivas na coluna, apresenta tronco também curto.
As pessoas com esta condição sofrem uma alteração ao nível das placas de crescimento que provoca também um aspeto nodoso nos ossos longos (metáfises) e causa um achatamento nas vértebras.
O que causa a Displasia Metatrópica?
A displasia metatrópica é causada por mutações no gene TRPV4. Este gene fornece instruções para a produção de uma proteína que atua como um canal de cálcio. Este canal é encontrado em muitos tipos de células, mas pouco se sabe sobre a sua função. Estudos sugerem que desempenha um papel no desenvolvimento normal da cartilagem e do osso1.
As mutações neste gene parecem sobreativar o canal de cálcio, aumentando o fluxo de iões de cálcio para as células. No entanto, mais estudos são necessários pois permanece pouco clara a relação entre as alterações na atividade do canal de cálcio e as caraterísticas específicas desta condição2.
Características Clínicas/Radiográficas3,4
Os sinais e sintomas desta condição podem variar entre leves e graves. Podem incluir o agravamento da curvatura da coluna (escoliose e cifose), o achatamento dos ossos da coluna vertebral (platispondilia) e uma restrição de certas articulações do corpo. Algumas pessoas nascem adicionalmente com um cóccix alongado (cauda coccígea).
As seguintes características são comuns entre as pessoas com displasia metatrópica:
-
Morfologia anormal do osso cortical
Alteração do osso cortical que forma a superfície densa dos ossos.
Frequência: muito frequente -
Ossificações condrais alteradas
Alteração do processo de ossificação endocondral que é um tipo de ossificação de substituição em que o tecido ósseo substitui a cartilagem.
Frequência: muito frequente -
Deformação dos corpos vertebrais
Morfologia anormal do corpo vertebral.
Frequência: muito frequente -
Deformação do disco intervertebral
Alteração estrutural do disco intervertebral.
Frequência: muito frequente -
Morfologia anómala das costelas
Frequência: muito frequente -
Alterações da metáfise
Alteração de uma ou mais metáfises, ou seja, da porção um pouco mais larga de um osso longo que é adjacente à placa de crescimento epifisário e cresce durante a infância.
Frequência: muito frequente -
Trabecularização metafisária grosseira
Aspeto irregular dos componentes da rede de tecido ósseo que compõe a estrutura celular do osso, ou seja, espessamento das linhas brancas (geralmente finas) que são produzidas por trabéculas em radiografias.
Frequência: muito frequente -
Ponte nasal baixa
Posicionamento posterior da raiz nasal em relação ao perfil facial habitual para a idade.
Frequência: muito frequente -
Pélvis em forma de alabarda
Aspeto radiográfico alterado da pélvis em desenvolvimento, em que a grande protuberância isquiática (ou tuberosidade isquiática) é pouco profunda e a pélvis assume o aspecto semelhante a uma alabarda (arma dos séculos XV e XVI).
Frequência: muito frequente -
Testa alta
Aumento da altura da testa.
Frequência: muito frequente -
Vértebras cervicais hipoplásicas
Vértebras cervicais subdesenvolvidas
Frequência: muito frequente -
Rigidez nas articulações
Sensação de aperto nas articulações ao tentar movê-las após um período de inactividade. A rigidez das articulações diminui normalmente com o tempo.
Frequência: muito frequente -
Cifose
Exagerada convexidade anterior da coluna vertebral torácica.
Frequência: muito frequente -
Tórax longo
Aumento da extensão inferior a superior do tórax.
Frequência: muito frequente -
Micromelia
Presença de extremidades pequenas.
Frequência: muito frequente -
Tórax estreito
Largura reduzida do peito de um lado para o outro, associada a uma distância reduzida do entalhe esternal até à ponta do ombro.
Frequência: muito frequente -
Escoliose
Presença de uma curvatura lateral anormal da coluna vertebral.
Frequência: muito frequente -
Estatura extremamente baixa
Grau severo de baixa estatura, mais de -4 SD da média corrigida por idade e sexo.
Frequência: muito frequente -
Aplasia/Hipoplasia dos pulmões
Pulmões ausentes/pouco desenvolvidos
Frequência: ocasional -
Camptodactilia
A articulação interfalângica distal e/ou a articulação interfalângica proximal dos dedos não pode ser estendida a 180 graus nem por extensão activa nem por extensão passiva.
Frequência: ocasional -
Catarata
Opacidade ou turvação que se desenvolve na lente cristalina do olho ou na sua cápsula.
Frequência: ocasional -
Fenda palatina
Defeito de desenvolvimento do palato resultante de uma falha de fusão dos processos palatinos e manifestando-se como uma separação do céu da boca (palato mole e palato duro).
Frequência: ocasional -
Clinodactilia do 5º dedo
Curvatura permanente do dedo mindinho
Frequência: ocasional -
Hidrocefalia
Líquido cerebrospinal em excesso no cérebro.
Frequência: ocasional -
Ouvidos baixos, com rotação posterior
Frequência: ocasional
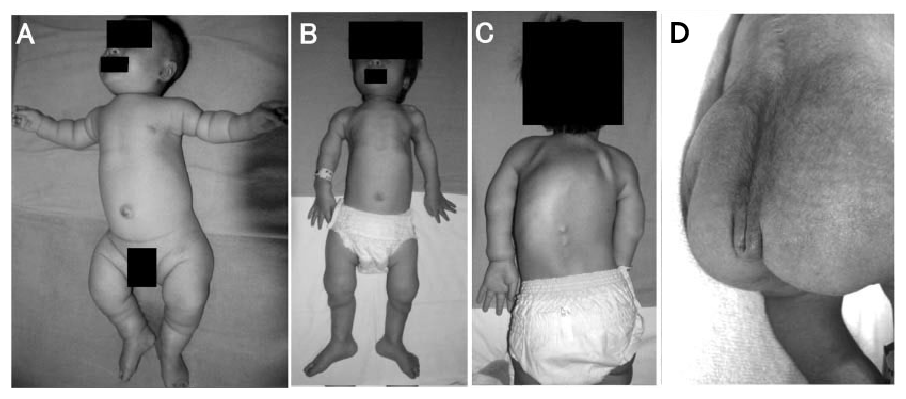
Fig. 1: Fotografias clínicas de recém-nascidos e crianças com displasia metatrópica
-
A: 4 meses. A criança mostra um tórax estreito, um tronco longo e membros curtos com dobras cutâneas redundantes.
-
B, C: 4 anos. A cifoscoliose severa desenvolve-se neste momento; no entanto, a proporção corporal ainda permanece micromélica. As grandes articulações estão expandidas.
-
D: Recém-nascido. Uma dobra dupla da pele é notada na região coccígea.
Fonte: Nishimura G, et al. 2012. TRPV4-associated skeletal dysplasias. Am J Med Genet Part C Semin Med Genet 160C:190–204.
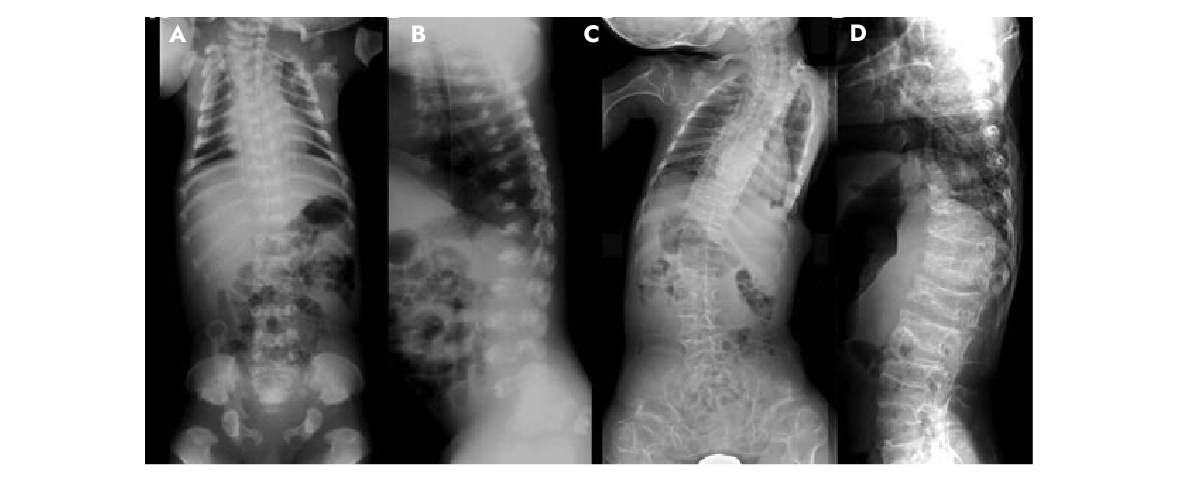
Fig. 2: Radiografias de recém-nascidos e crianças com displasia metatrópica
-
A,B: Recém-nascidos. C,D: 3 e 6 anos.
-
No período neonatal e na infância, os corpos vertebrais são finos, com largos espaços intervertebrais e sincondrose neurocentral muito ampla.
-
Os arcos neurais posteriores são volumosos. Com o tempo, a ossificação vertebral progride e os corpos vertebrais tornam-se alongados.
-
A cifoescoliose é progressiva. As extremidades anteriores das costelas tornam-se proeminentes com a idade.
Fonte: Nishimura G, et al. 2012. TRPV4-associated skeletal dysplasias. Am J Med Genet Part C Semin Med Genet 160C:190–204.
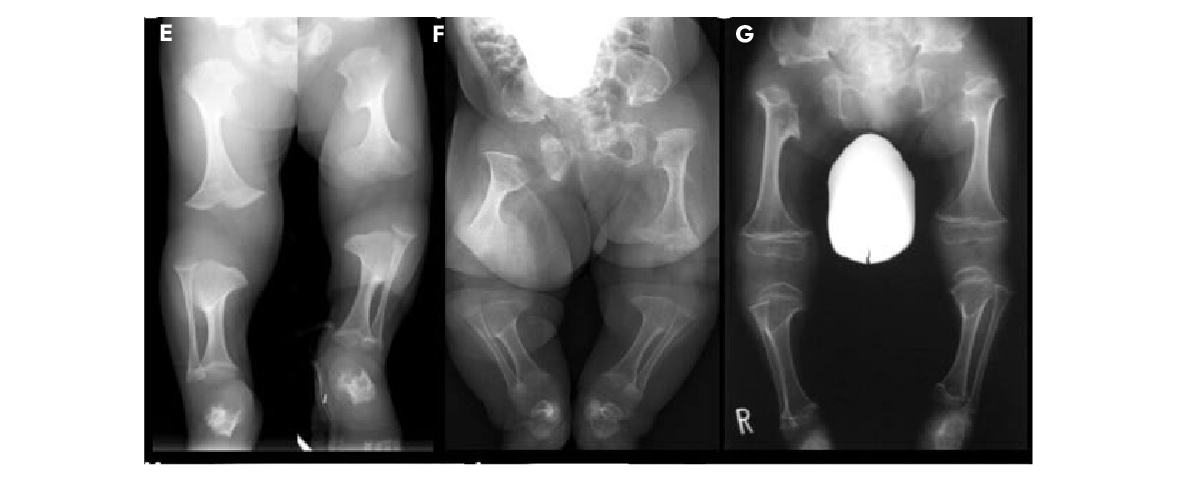
Fig. 3: Radiografias de recém-nascidos e crianças com displasia metatrópica
-
E: Recém-nascido F: 3 meses. G: 3 anos.
-
Alteração dos ossos longos muito mais severa em idade mais jovem. Na infância, as metáfises dos ossos longos tornam-se côncavas e circundadas por um colar metafisário esclerótico.
-
Os ossos tubulares curtos são finos no período neonatal, mas tornam-se mais grossos com a idade.
Fonte: Nishimura G, et al. 2012. TRPV4-associated skeletal dysplasias. Am J Med Genet Part C Semin Med Genet 160C:190–204.
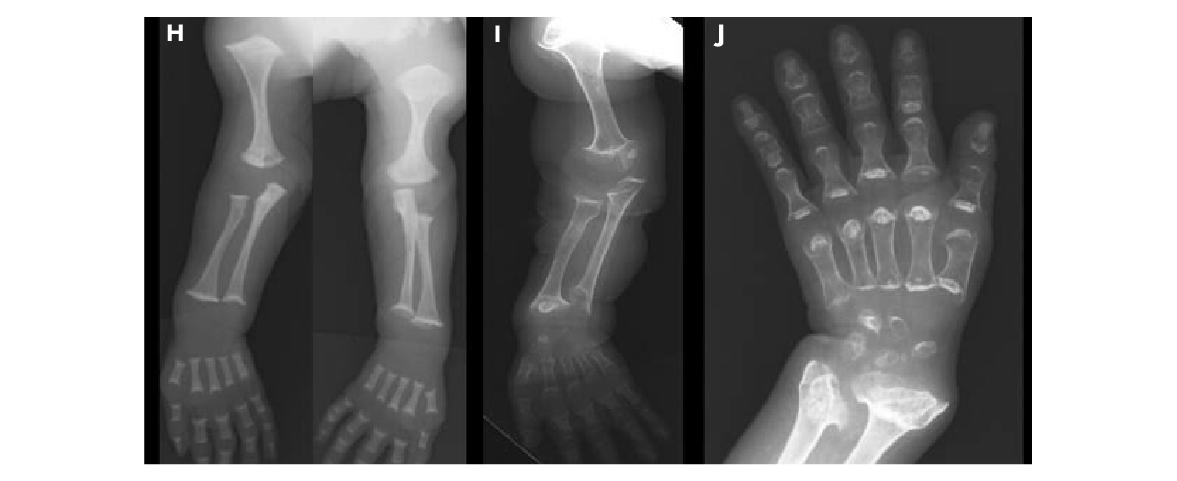
Fig. 4: Radiografias de crianças com displasia metatrópica
-
H: Recém-nascido. I: 2 anos. J: 5 anos.
-
Na infância, a ossificação epifisária está atrasada em todos os ossos longos. Com o avançar da idade, as epífises da maioria dos ossos longos ficam bastante desenvolvidas e muito grandes, mas as epífises proximais do fémur continuam sem ossificação.
-
A ossificação do tarso é muito irregular no período neonatal, como em (E).
Fonte: Nishimura G, et al. 2012. TRPV4-associated skeletal dysplasias. Am J Med Genet Part C Semin Med Genet 160C:190–204.
Diagnóstico4
O diagnóstico baseia-se na verificação das características clínicas e radiológicas referidas acima. Os achados radiológicos incluem diáfises curtas com metáfises largas, platispondilia marcada, calcificação precoce da cartilagem hióide e cricóide, pelve em forma de alabarda, hipoplasia grave anterior das primeiras vértebras cervicais e ossos do calcanhar irregulares (quadrados).
Testes Genéticos
Uma vez que as manifestações radiológicas mudam consoante a idade, os testes genéticos podem identificar uma mutação no gene TRPV4, confirmando o diagnóstico. Estes permitem também uma confirmação pré-natal da condição.
Diagnóstico Pré-Natal
O diagnóstico pré-natal é possível através de testes genéticos e avaliação pré-natal, com uma tomografia computorizada em 3D ou uma ultrassonografia (ecografia), e deve ser recomendado quando é conhecida uma mutação na família.
Estes exames de imagem podem mostrar sinais da condição, incluindo um peito estreito e uma aparência de haltere nos ossos longos.
Ultrassonografia
A ultrassonografia é a principal forma de diagnóstico através de imagens quando há suspeita de displasia óssea pré-natal.
Diagnóstico diferencial:
- Mucopolissacaridose tipo IV ou síndrome de Morquio A
- Displasia espondilometafisária, em particular a de tipo Kozlowski.
Tratamento5,6
O tratamento da displasia metatrópica é direcionado para os sintomas específicos de cada pessoa e pode exigir os cuidados coordenados de uma equipa de especialistas, onde se incluem pediatras, ortopedistas, cirurgiões, fisioterapeutas, oftalmologistas e outros profissionais de saúde.
Alguns exemplos de tratamentos:
-
Para a instabilidade cervical: fusão e descompressão da coluna vertebral se houver estenose presente, bem como a possível colocação de uma auréola
-
Para o bloqueio das vias respiratórias: traqueostomia e apoio ventilatório a longo prazo podem ser necessários em casos graves.
-
Para a escoliose: o tratamento pode incluir uma fusão espinal
-
Para fraturas do calcanhar: o osso do calcanhar é como um ovo, com casca forte e interior macio e pode estilhaçar quando é traumatizado. O tratamento requer reparação de múltiplas fracturas, bem como a restauração da articulação subtalar. Se a fractura não tiver deslocado o osso, o repouso e a imobilização parcial podem resultar. Algumas fraturas do calcanhar podem ser tratadas sem cirurgia, através da manipulação do pé enquanto a pessoa está sob anestesia. Por vezes, a cirurgia pode ser necessária em casos graves. Se a articulação subtalar for gravemente danificada, a fusão é a única opção
Aconselhamento Genético
O aconselhamento genético será benéfico para as pessoas com displasia metatrópica e para as suas famílias. Fornece informações sobre a natureza, hereditariedade e implicações das condições genéticas, ajudando a tomar decisões médicas e pessoais informadas, essenciais, por exemplo, para o planeamento familiar, onde se deve esclarecer sobre o risco genético e a possibilidade de testes pré-natais.
É apropriado oferecer aconselhamento genético (incluindo a discussão de potenciais riscos para os descendentes e opções reprodutivas) aos jovens adultos com mutação.
Bibliografia
- Genevieve, D., et al. Long-term follow-up in a patient with metatropic dysplasia. (Letter) Am. J. Med. Genet. 135A: 342-343, 2005.
- Camacho, N., et al. Dominant TRPV4 mutations in nonlethal and lethal metatropic dysplasia. Am. J. Med. Genet. 152A: 1169-1177, 2010.
- Nishimura G, et al. 2012. TRPV4-associated skeletal dysplasias. Am J Med Genet Part C Semin Med Genet 160C:190–204.
- Dai, J., et al. Novel and recurrent TRPV4 mutations and their association with distinct phenotypes within the TRPV4 dysplasia family. J. Med. Genet. 47: 704-709, 2010.
- cedars-sinai.org - consultado no dia 11 de julho de 2022
- orpha.net - consultado no dia 11 de julho de 2022
A ANDO gostaria de agradecer à família das irmãs Miriam e Jéssica (na fotografia principal desta página, em cima) pela amável cedência da imagem.
Partilhe a sua história
Gostaria de partilhar a sua experiência? Pode fazê-lo na primeira pessoa ou enquanto mãe, pai ou cuidador. O seu testemunho pode ajudar a informar, sensibilizar e inspirar outras pessoas.
partilhar a minha história