
Hipofosfatásia
A hipofosfatásia ou HPP é uma condição rara metabólica caraterizada que causa alterações nos ossos e dentes. Não existem dados exactos de prevalência e incidência da hipofosfatásia. No entanto, no Norte e Oeste da Europa, a prevalência das formas graves desta condição (perinatais letais e infantis) foi estimada em 1/300 000. Calcula-se que as formas moderadas sejam mais frequentes com uma prevalência de 1/63001.
Veja o vídeo da AERyOH (associação espanhola) que explica o que é a HPP:
O que é a Hipofosfatásia?
A hipofosfatásia é uma forma genética rara de raquitismo ou osteomalácia. que apresenta, paradoxalmente, baixa atividade plasmática da fosfatase alcalina (ALP). A sua gravidade varia com as consequências clínicas. Pode causar morte in utero de um esqueleto desmineralizado ou pode apenas provocar problemas nos dentes durante a vida adulta.
A idade na qual as características clínicas se tornam aparentes distingue as formas perinatal, infantil (antes dos 6 meses), juvenil (depois dos 6 meses) e adulta da doença. Pessoas que apresentam apenas sintomas dentários têm odonto-HPP.
O que causa a Hipofosfatásia
São conhecidas mais de 400 mutações diferentes no gene ALPL que causam hipofosfatásia. Este gene dá instruções para a produção de uma enzima chamada fosfatase alcalina, isozima não específica do tecido (TNSALP) que está envolvida na mineralização dos ossos e dos dentes. As mutações neste gene podem criar uma versão anómala dessa enzima, afetando assim o processo de mineralização e conduzindo às características da HPP.
As mutações que eliminam quase completamente a atividade da fosfatase alcalina causam, geralmente, as formas mais severas de HPP, enquanto que as mutações que reduzem a atividade em menor grau provocam as formas mais suaves de HPP2.
Características Clinicas
Os sinais e sintomas da HPP variam muito e podem aparecer em qualquer momento, desde antes do nascimento até à idade adulta:
Hipofosfatásia Perinatal
-
Membros curtos e deformados
-
Caput membranaceum da hipomineralização esquelética profunda
-
Esporões osteocôndricos incomuns podem perfurar a pele e sair lateralmente no eixo médio das ulnas e fíbulas.
-
Pode haver um choro com tom alto e irritabilidade
-
Apneia periódica com cianose e bradicardia
-
Febre inexplicada
-
Anemia
-
Hemorragia intracraniana
Alguns recém-nascidos vivem alguns dias, mas sofrem comprometimento respiratório crescente dos defeitos no tórax e pulmões hipoplásicos. Muito raramente há sobrevivência a longo prazo.
Hipofosfatasia Pré-natal Benigna
-
Manifestação de alterações in utero, mas cujos cursos pós-natais apresentaram melhoria esquelética espontânea.
Hipofosfatásia infantil
-
Falha de crescimento
-
Hipotonia
-
Raquitismo
-
Convulsões
-
Ossificação diminuída do crânio
-
Pode ocorrer protrusão da fontanela anterior, pressão intracraniana elevada com papiledema, proptose e braquicefalia
-
Escleras azuladas
-
Tórax instável que predispõe pneumonia pode ocorrer pela deformidade raquítica do tórax e fraturas da costela
-
Fraqueza e metas motoras atrasadas
-
Epilepsia dependente de vitamina B6
-
Fusão óssea verdadeira das suturas cranianas pode ocorrer prematuramente
-
Hipercalcemia e hipercalciúria podem causar vômitos recorrentes, nefrocalcinose, e comprometimento renal
-
Craniossinostose funcional.
Hipofosfatásia juvenil
-
Perda prematura de dentes decíduos (antes dos 5 anos)
-
Formação de contas na junção costocondral
-
Pernas curvadas ou genu valgum
-
Alargamento dos pulsos, joelhos e tornozelos
-
Crânio braquicefálico
-
Baixa estatura e atraso na marcha
-
Pode ocorrer dor esquelética e rigidez, assim como episódios de desconforto articular e inchaço.
Hipofosfatásia Adulta
-
Dor nos pés causada por fraturas recorrentes e mal cicatrizadas do metatarso
-
Desconforto nos quadris ou coxas devido a pseudofraturas femorais
-
Fraturas espalhadas e não cicatrizadas podem então causar debilidade significativa
-
Perda precoce ou extração da dentição
-
Deposição de pirofosfato di-hidrato de cálcio (CPPD) que pode causar artropatia PPi, e, ocasionalmente, pseudogota
-
Deposição paradoxal de cristais de hidroxiapatitae
-
Ossificação dos ligamentos (hiperostose espinhal)
-
Hiperparatireoidismo primário (raramente)
-
Osteomalácia, osteopenia generalizada e condrocalcinose, e, por vezes, fraturas de artropatia PPi ou periartrite por calcificação.
Odonto-HPP
A forma mais leve de HPP é diagnosticada quando a única anormalidade clínica aparente é doença dentária. Aqui, não há evidência radiográfica ou por biópsia óssea de doença esquelética por HPP.
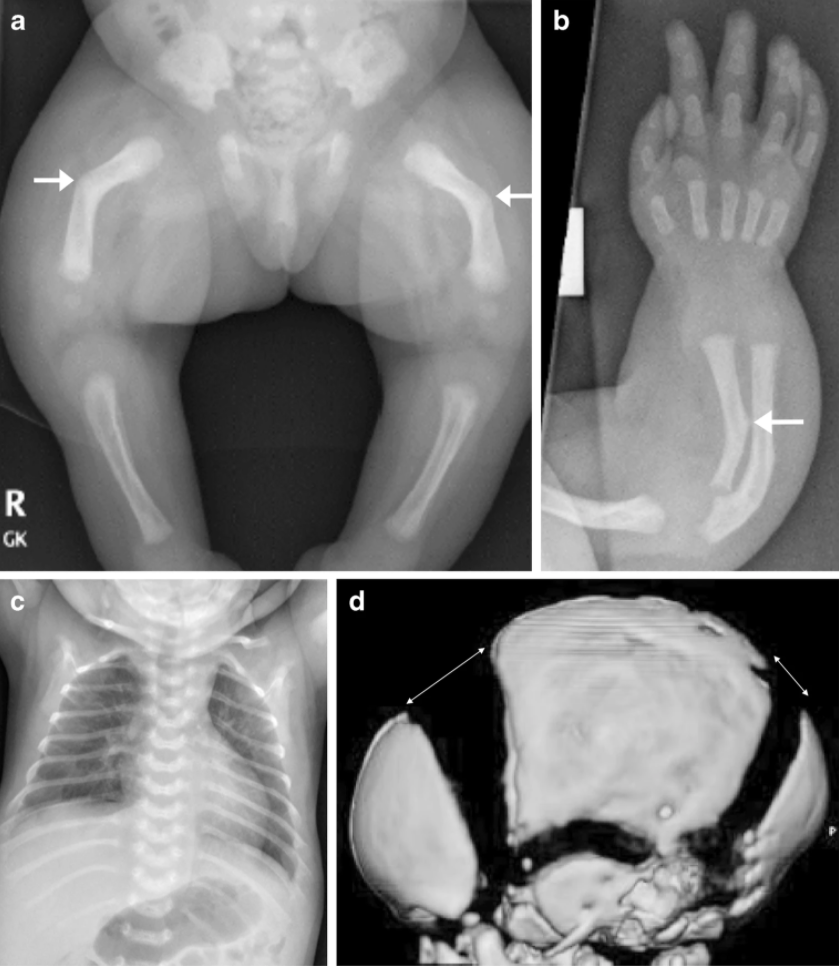 |
|
Fig.1. HPP numa menina de 3 meses. a-c) Radiografias mostram femur de arco curto (a). Cúbito com raio de esporão do antebraço direito (b) e costelas finas e mal ossificadas (c). TAC mostra suturas largas e fontanelas (d). Fonte: Radcliffe Publishing, London4 |
Tratamento
Até há pouco tempo, a gestão da HPP tinha como principal objectivo atenuar os sintomas da condição:
-
Hidratação
-
Restrição de cálcio dietético, vitamina D, e por vezes diuréticos de tiazida para hipercalcemia
-
Apoio ventilatório para bebés gravemente afectados, alguns dos quais necessitam de uma traqueostomia
-
Fisioterapia, terapia ocupacional e gestão da dor crónica
-
Cirurgia para fracturas que não cicatrizam.
Para crianças, o tratamento consiste na administração oral sistemática de fosfato e calcitriol (associados à monitorização frequente da altura), cálcio sérico, hormona paratiróide, concentrações séricas de fosfato, e cálcio urinário e creatinina. O calcitriol ajuda a prevenir o hiperparatiroidismo secundário que pode ser induzido pela administração de fosfato. Na idade adulta, a terapia é geralmente continuada em doentes sintomáticos e visa geralmente reduzir a dor óssea. A cirurgia correctiva de deformidades esqueléticas pode ser necessária em alguns casos. Por vezes, a nefrocalcinose e o hiperparatiroidismo podem ser observados como complicações da terapia; por conseguinte, é necessário um acompanhamento frequente.
Tratamento Aprovado pela EMA/FDA
A Asfotase alfa (comercializada com o nome Strensiq™) foi designada Medicamento Órfão (um medicamento utilizado em doenças raras) pela Agência Europeia do Medicamento para o tratamento da HPP a 3 de Dezembro de 2008. Trata-se de uma versão da enzima fosfatase alcalina que atua substituindo-a no organismo, estando por isso indicada para a terapia de substituição enzimática a longo prazo em doentes com HPP pediátrica, para tratamento das suas alterações ósseas5.
Bibliografia
1. Orphanet
2. Hypophosphatasia. Genetics Home Reference (GHR). jan. 2021; http://ghr.nlm.nih.gov/condition=hypophosphatasia
3. Hipofosfatásia: Uma Visão geral para Médicos e Professionais Médicos. Soft Bones Inc., The U.S. Hypophosphatasia Foundation. 2017
4. Hall CM, et al (2012) Diagnosis of fetal skeletal dysplasias. In: Fetal and perinatal skeletal dysplasias: an atlas of multimodality imaging. Radcliffe Publishing, London
5. EMA. Strensiq, Asfotase alfa. https://www.ema.europa.eu/en/medicines/human/EPAR/strensiq
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
Doenças raras e displasias ósseas com os nossos vídeos informativos
Partilhe a sua história
Gostaria de partilhar a sua experiência? Pode fazê-lo na primeira pessoa ou enquanto mãe, pai ou cuidador. O seu testemunho pode ajudar a informar, sensibilizar e inspirar outras pessoas.
partilhar a minha história