
Condrodisplasia Metafisária Tipo Schmid
A Condrodisplasia Metafisária tipo Schmid (CMtS) é uma displasia condrometafisária muito rara com incidência e prevalência desconhecidas. Origina alterações ósseas que se concentram na região metafisária dos ossos longos, baixa estatura com encurtamento e curvatura dos membros por alteração da função normal da placa de crescimento1.
O que é a CMtS1,2?
A condrodisplasia metafisária de tipo Schmid é um tipo de condrodisplasia associada a uma deficiência de colagénio, tipo X. Ao contrário de outras "síndromes de raquitismo", os indivíduos afectados têm níveis séricos normais de cálcio, fósforo e aminoácidos urinários. Os ossos longos são curtos e curvos, com placas de crescimento e metáfises alargadas. O seu nome deve-se ao investigador alemão F. Schmid, que a caracterizou em 1949. A Condrodisplasia Metafisária tipo Schmid apresenta características físicas que podem incluir protuberância dos ossos da caixa torácica inferior, lordose lombar, dor nas pernas, e/ou alterações da anca em que o osso da coxa é inclinado para o centro do corpo (coxa vara). Estas alterações podem manifestar-se ao nascimento ou após o primeiro ano de vida, e mais tarde levam ao surgimento de uma marcha invulgar.
O que causa a CMtS2?
A CMtS é causada por uma mutação autossómica dominante do gene do colagénio tipo X chamado COL10A1. Logo, para se manifestar, basta ser transmitido uma cópia de um gene mutado. Este pode ser herdado de qualquer um dos progenitores, ou pode ser o resultado de uma nova mutação, chamada mutação de novo. O risco de passar o gene com mutação do progenitor com mutação para a descendência é de 50% para cada gravidez. (ver imagem abaixo).
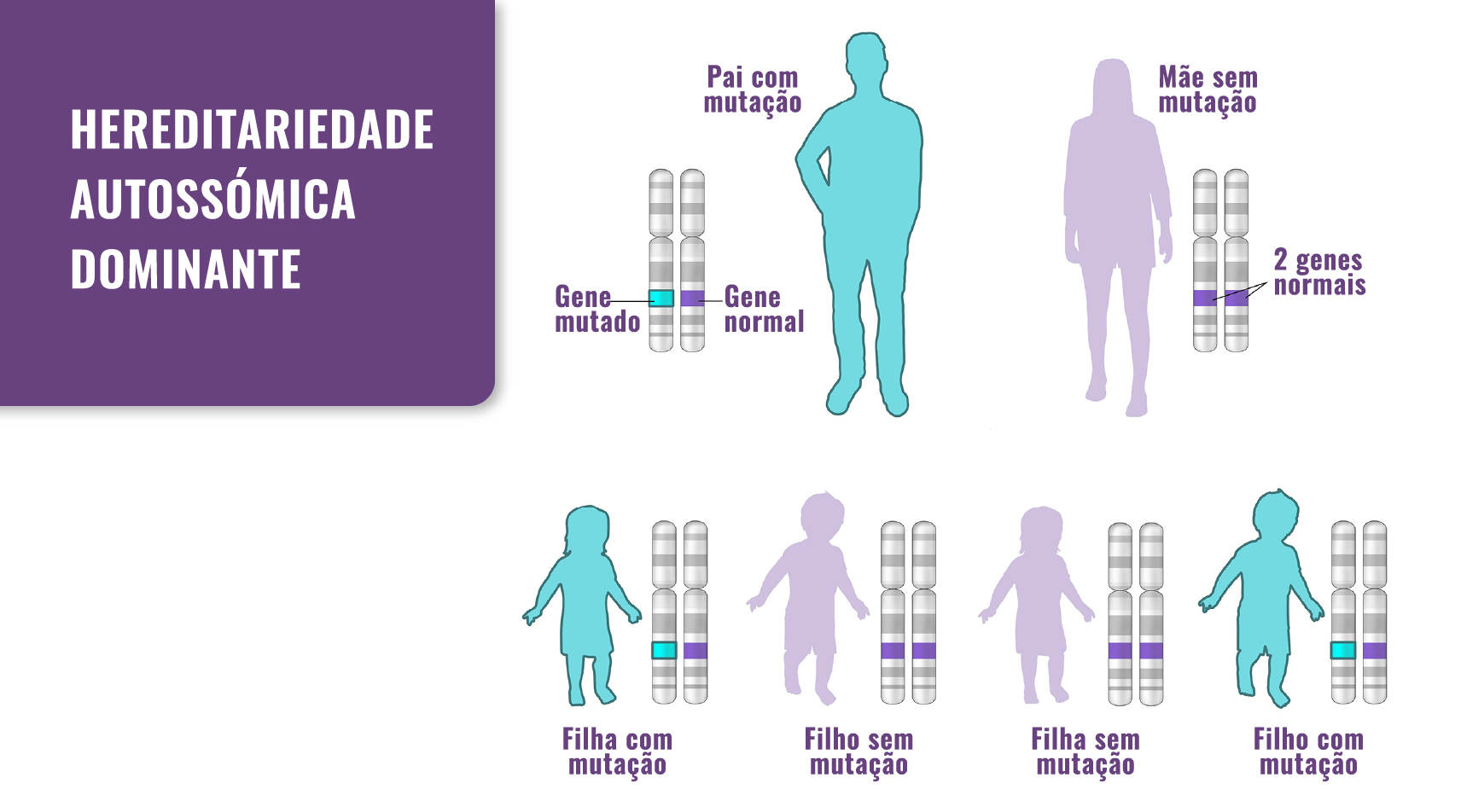
Fig. 1. Hereditariedade: a CMtS passa de pais para filhos. No entanto, também pode ocorrer como resultado de uma mutação de novo.
Características Clínicas e Radiológicas1,3
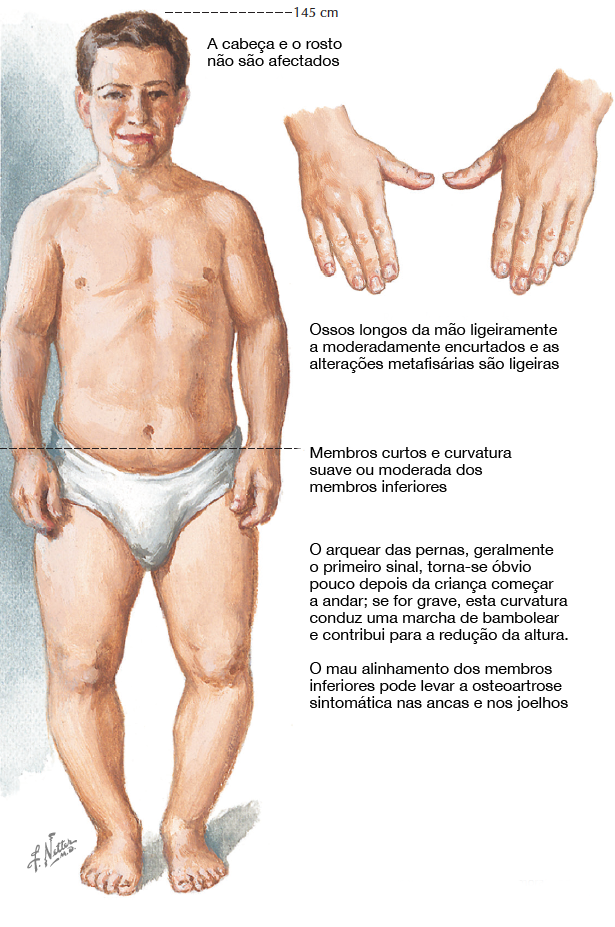 Fig. 2. Características comuns da CMtS. Créditos: Netter.
Fig. 2. Características comuns da CMtS. Créditos: Netter.
As características clínicas e radiográficas não estão habitualmente presentes no nascimento, mas manifestam-se na primeira infância:
-
Estatura progressivamente baixa (a partir dos dois anos de idade)
-
Membros curtos
-
Genu varum, coxa vara e tornozelo varo
-
Marcha bamboleante
-
Face e cabeça de aspeto normal
-
As radiografias mostram irregularidades metafisárias dos ossos longos
-
Ossos tubulares curtos
-
Esclerose das costelas
-
Concavidade metafisária dos metacárpicos e falanges proximais
-
Dores nas articulações dos joelhos e ancas são comuns e podem limitar a actividade física
-
Não ocorrem manifestações extraesqueléticas
-
A estatura em adulto pode chegar aos 150 cm
-
Na idade adulta, podem aparecer sinais de artrose.
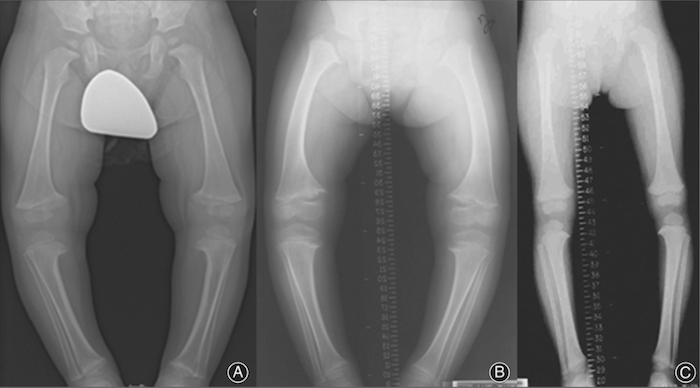 |
| Fig.3. A: Genu varum em rapaz de 5 anos com curvatura das pernas; B: genu varum em rapariga de 7 anos; C: genu varum menos pronunciado em rapariga de 9 anos. Todas as crianças mostraram um alargamento das placas de crescimento associada à concavidade metafisária, que se assemelha ao raquitismo. Fonte: Wiley Online Library |
Diagnóstico1
O diagnóstico da CMtS é estabelecido através da deteção dos elementos clínicos e radiográficos característicos e/ou recorrendo à identificação de uma variante patogénica heterozigótica em COL10A1 através de testes genéticos moleculares.
Tratamento1,3
Os tratamentos da CMtS direcionam-se para a gestão das complicações ortopédicas. A fisioterapia e/ou cirurgia ortopédica pode ajudar a corrigir alterações específicas, tais como a deformidade da anca. É importante uma intervenção precoce para assegurar que as crianças alcançam o seu potencial de mobilidade assim como apoio por terapias específicas como terapia da fala, fisioterapia, e outros serviços médicos, sociais e/ou vocacionais.
É recomendada uma avaliação anual do crescimento, avaliação clínica das manifestações ortopédicas e avaliação psicossocial. O aconselhamento genético será também benéfico para os indivíduos afectados e as suas famílias.
Tratamentos em Investigação
A carbamazepina é um medicamento aprovado pela FDA e recomendado para aprovação pela EMA para uso em epilepsia e desordem afectiva bipolar. A sua acção adicional como estimulador das vias de autofagia e de degradação proteasomal conduziu ao pedido de nova indicação como tratamento para a Condrodisplasia Metafisária Tipo Schmid.
Para mais informações sobre a carbamazepina no tratamento da Condrodisplasia Metafisária Tipo Schmid consulte a nossa página dedicada a este tema, aqui.
Para mais informação sobre tratamentos emergentes e sobre o processo de inovação e desenvolvimento de medicamentos para as doenças raras, consulte a nossa página dedicada à investigação para as Displasias Ósseas aqui.
Bibliografia
- 1. Al Kaissi A, et al. Schmid's type of metaphyseal chondrodysplasia: diagnosis and management. Orthop Surg. 2018;10:241–6
- 2. Bateman JF, Wilson R, Freddi S, Lamandé SR, Savarirayan R. Mutations of COL10A1 in Schmid metaphyseal chondrodysplasia. Hum Mutat. 2005;25:525–34
- 3. Pinto J et al. "Estudo da displasia condrometafisári do tipo Schmid", 1994
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui
-
doenças raras e displasias ósseas através dos nossos vídeos informativos