
Displasia Espondiloepimetafisária Tipo Strudwick
A Displasia Espondiloepimetafisária tipo Strudwick (SEMD tipo Strudwick) é uma condição hereditária do crescimento ósseo que apresenta estatura desproporcionalmente baixa desde o nascimento. É uma condição extremamente rara e até hoje foi identificada em menos de 30 pessoas.
O que é a SEMD tipo Strudwick1?
A SEMD tipo Strudwick é uma displasia óssea ultra rara com transmissão autossómica dominante, pertencente ao grupo das displasias com orgigem nas alterações do gene COL2A1. As pessoas apresentam um tronco e membros muito curtos, alterações esqueléticas (lordose, escoliose, vértebras achatadas, pectus carinatum, coxa vara, pé torto e epífises ou metáfises anormais) e problemas de visão. O nome Strudwick provém da primeira pessoa com esta doença.
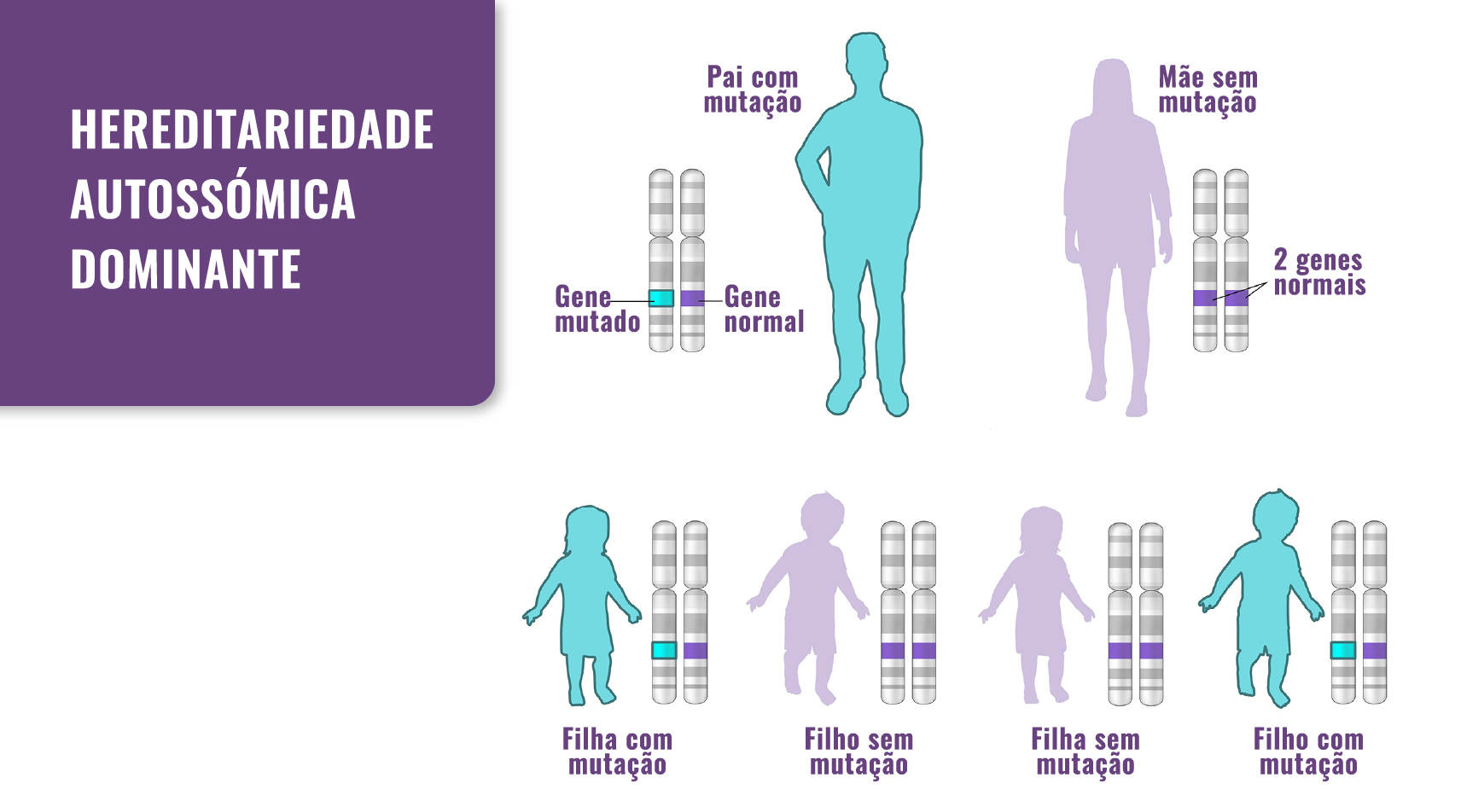
Fig. 1. Hereditariedade: a SEMD tipo Strudwick passa de pais para filhos. No entanto, também pode ocorrer como resultado de uma mutação de novo.
O que causa a SEMD tipo Strudwick1?
A SEMD tipo Strudwick é herdada num padrão autossómico dominante, o que significa que uma cópia do gene alterado em cada célula é suficiente para a causar.
Esta condição pertence a um espectro de perturbações esqueléticas causadas por mutações no gene COL2A1 que fornece instruções para a produção de uma proteína que, por sua vez forma o colagénio tipo II. Este tipo de colagénio encontra-se principalmente no gel transparente que preenche o globo ocular (o vítreo) e na cartilagem. A cartilagem é um tecido resistente e flexível que compõe grande parte do esqueleto durante o seu desenvolvimento inicial. A maior parte da cartilagem é posteriormente convertida em osso, excepto a cartilagem que continua a cobrir e proteger as extremidades dos ossos e que está presente no nariz e nas orelhas externas. O colagénio de tipo II é essencial para o desenvolvimento normal dos ossos e outros tecidos conjuntivos que formam o quadro de suporte do corpo.
Características Clínicas e Radiológicas2
-
Estatura baixa desde o nascimento, com um tronco muito curto
-
Aplasia/hipoplasia envolvendo ossos das extremidades
-
Metáfises expandidas
-
Vértebras anormalmente ossificadas
-
Instabilidade cervical (aumenta o risco de danos na medula espinal)
-
Face plana
-
Glossoptose (retracção da língua)
-
Problemas auditivos
-
Hipertelorismo (olhos afastados)
-
Pévis hipoplásica
-
Micrognatia (maxilar inferior pequeno)
-
Problemas oftalmológicos que podem prejudicar a visão são comuns (miopia severa e descolamento da retina).
-
Encurtamento de ossos longos
-
Pequenas epífises
-
Lordose e escoliose (podem ser graves e causar problemas respiratórios)
-
Platispondilia (vértebras achatadas)
-
Pectus carinatum (protrusão grave do osso do peito - ver Fig. 1)
-
Pé torto (Fig. 2)
-
Coxa vara (Alterações na articulação da anca que faz com que os ossos se virem para dentro)
-
Pé torto
-
A artrose pode desenvolver-se no início da vida
-
Palato fendido
 |
|
Fig 2. Pessoa com pectus carinatum em que o osso do peito é empurrado para fora. Créditos: Credit: GeneReviews ©1993-2020 University of Washington |
|
|
|
Fig 3. Bebé com pé torto. Créditos: Alila Medical Media/Shutterstock.com |

Diagnóstico
Realizar um diagnóstico para uma doença genética ou rara pode muitas vezes ser um desafio. Os profissionais de saúde normalmente analisam o historial médico e os sintomas de uma pessoa, efetuam um exame físico e testes laboratoriais a fim de obter um diagnóstico.
Diagnóstico Diferencial3,4,5
Os bebés com SEMD tipo Strudwick, apresentam inicialmente as mesmas características clínicas e radiográficas que os bebés com SEDC. No entanto, no primeiro ano de vida, as características metafisárias permitem distinguir as duas condições, sugerindo o diagnóstico. A evolução clínica é semelhante à da SEDC, mas com risco acrescido de instabilidade cervical e compressão da medula espinal, o que representa o maior risco para estes indivíduos3,4.
O diagnóstico diferencial das colagenopatias tipo II (onde se inclui a SEMD tipo Strudwick) envolve uma série de perturbações, desde displasias esqueléticas graves, frequentemente letais, com ossificação anormal e grandes anomalias esqueléticas, até condições mais suaves com resultados clínicos e radiográficos limitados. As perturbações de etiologia genética conhecida e desconhecida a considerar no diagnóstico diferencial das colagenopatias tipo II poderão ser encontradas aqui5.
Tratamento6
Não existe tratamento para a causa da SEMD tipo Strudwick e as terapias específicas utilizadas servem para tratar ou atenuar os sintomas. Os médicos devem monitorizar cuidadosamente os bebés de forma a assegurar uma detecção imediata e uma prevenção ou tratamento correctivo adequado das dificuldades que vão surgindo.
O tratamento dos sintomas pode incluir:
- Gestão cirúrgica para a medulopatia (fixação C1-C2) e para a escoliose severa e progressiva
- Ventilação mecânica e cirurgia do palato fendido (no caso de problemas respiratórios)
- A miopia deve ser corrigida com o uso de lentes.
- As pessoas em risco devem ser informadas sobre sinais e sintomas de descolamento da retina.
- Para os problemas articulares (laxismo, contraturas e dor devido a artrose precoce): fisioterapia, analgésicos para dor e avaliação por parte de ortopedista.
De acordo com as necessidades de cada pessoa podem surgir outros sintomas que necessitem de outras terapias. O acompanhamento deve ser feito por uma equipa de especialistas: pediatras, ortopedistas, oftalmologistas, reumatologistas, fisioterapeutas e outros profissionais de saúde.
Estudos a Decorrer
Está a decorrer um estudo observacional da história natural em crianças com colagenopatias de tipo II e baixa estatura (incluindo a SEMD tipo Strudwick) que pode ser consultado aqui.
Bibliografia
1. Tiller, G. E., et al. Dominant mutations in the type II collagen gene, COL2A1, produce spondyloepimetaphyseal dysplasia, Strudwick type. Nature Genet. 11: 87-89, 1995. PubMed: 7550321
2. Amirfeyz R, et al. Orthopaedic manifestations and management of spondyloepimetaphyseal dysplasia Strudwick type. J Pediatr Orthop B. 2006 Jan;15(1):41-4.
3. Walter K, et al. COL2A1-related skeletal dysplasias with predominant metaphyseal involvement. Am J Med Genet A. 2007;143A:161–7.
4. Terhal PA, et al. A study of the clinical and radiological features in a cohort of 93 patients with a COL2A1 mutation causing spondyloepiphyseal dysplasia congenita or a related phenotype. Am J Med Genet A. 2015;167A:461–75.
5. Gregersen PA, Savarirayan R. Type II Collagen Disorders Overview. 2019 Apr 25. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle (WA): University of Washington; 1993-2021. Available from: https://www.ncbi.nlm.nih.gov/books/NBK540447/
6. Savarirayan R, et al. Best practice guidelines regarding diagnosis and management of patients with type II collagen disorders. Genet Med. 2019. Epub ahead of print.
Saiba mais sobre:
-
O processo de inovação e desenvolvimento (I&D) de medicamentos para doenças raras
-
A Dor nas displasias ósseas aqui
-
doenças raras e displasias ósseas através dos nossos vídeos informativos
Partilhe a sua história
Gostaria de partilhar a sua experiência? Pode fazê-lo na primeira pessoa ou enquanto mãe, pai ou cuidador. O seu testemunho pode ajudar a informar, sensibilizar e inspirar outras pessoas.
partilhar a minha história